Auf dem DGN gesehen: Akte ungelöst – Neurologen decken auf!
Beim Sanofi Genzyme-Symposium "Akte ungelöst - Neurologen decken auf!" wurden auf dem diesjährigen Kongress der Deutschen Gesellschaft für Neurologie in Leipzig ebensolche Fälle vorgestellt.
- Muskelgewebsentzündungen, Muskelschwächen und/oder Zwerchfell- bzw. Atemschwäche können Folge einer Vielzahl von neuromuskulären Erkrankungen sein.
- Treten proximal betonte Muskelschwächen in Verbindung mit moderater Hyper-CK-ämie auf, sollte auch an die seltene lysosomale Glykogen-Speicherkrankheit Morbus Pompe gedacht werden.
- Eine frühe Diagnose ermöglicht eine frühzeitige kausale Therapie.
- Die Wirksamkeit und Bedeutung der seit 11 Jahren verfügbaren Enzymersatztherapie wurde mit den aktuell publizierten Empfehlungen des Europäischen Pompe Consortiums (EPOC) zur EET bestätigt2.
Bei seltenen oder weniger bekannten Krankheitsbildern fällt die Diagnose oft schwer. Es dauert in vielen Fällen Jahre, bis die richtige Diagnose gestellt und für die Patienten Gewissheit erreicht wird. Häufig wird bei der Diagnosefindung ein interdisziplinäres Konzil einberufen, das über die Grenzen eines Fachbereichs hinaus versucht, Ursachen von Beschwerden zu ergründen.
Beim Sanofi Genzyme-Symposium "Akte ungelöst - Neurologen decken auf!" wurden auf dem diesjährigen Kongress der Deutschen Gesellschaft für Neurologie in Leipzig ebensolche Fälle vorgestellt.
Der erste Vortrag ("Nicht wasserscheu und doch Angst vorm Schwimmen?") von Prof. Dr. Peter Young von der Klinik für Schlafmedizin und Neuromuskuläre Erkrankungen am Universitätsklinikum Münster handelte weder vom Schwimmunterricht, noch von psychosomatisch bedingten Angstzuständen. Vielmehr ging es um zwei Patientenfälle mit recht ähnlichen Symptomen wie zunehmender Atemnot, insbesondere beim Schwimmen (mit ausgeprägter Angstreaktion), und einer erkennbaren Zwerchfellschwäche.
Der zuerst vorgestellte Patient berichtet neben dem Genannten von einer Schmerzsymptomatik (Neuralgie) im linken Arm, Schulter und entlang der Wirbelsäule. Die Untersuchung zeigt eine Parese der handstreckenden Muskeln links. Es wurden bereits ein Pneumologe und ein Neurologe aufgesucht. Die Diagnose blieb unklar, eine Lungenerkrankung konnte ausgeschlossen werden.
Zur weiteren diagnostischen Abklärung wurde der Patient ins Schlaflabor geschickt. Hier wurden eine Zwerchfellunbeweglichkeit und eine REM-assoziierte Hypoventilation festgestellt.
Youngs Frage ans Publikum, ob es sich hier um eine oder zwei Erkrankungen handelt, wurde vergleichsweise scheu beantwortet: ein Schlaglicht auf die vielen Unsicherheiten und offenen Fragen, vor die sich Mediziner in der Diagnose seltener Krankheitsbilder immer wieder gestellt sehen.
Auch der zweite Patient zeigte eine zunehmende Atemnot beim Schwimmen, später auch beim Treppensteigen, sonst bemerkte er keine verminderte körperliche Leistungsfähigkeit. Der Patient wurde mehrfach bei verschiedenen Pneumologen ohne klares Diagnoseergebnis vorstellig. Beim Hausarzt wurden erhöhte Kreatinkinase (CK)-Werte (500-1000 U/ml) gemessen. Weitere Untersuchungen zeigten im Liegen ein paradoxes Atemmuster bei inspiratorisch einfallender Bauchdecke, einen sehr schwachen Hustenstoß sowie im Schlaflabor eine Hypoventilation mit Hyperkapnie im REM-Schlaf.
Die schließlich enthüllten Diagnosen (Parsonage-Turner-Syndrom bei Fall 1 und die seltene lysosomale Glykogen-Speicherkrankheit Morbus Pompe bei Fall 2) basieren, neben einigen anderen Leitsymptomen, auf der Identifikation einer Zwerchfellschwäche (eine restriktive Ventilationsstörung mit Hypoventilation), welche auch für die paradoxen Atmungsmuster verantwortlich waren.
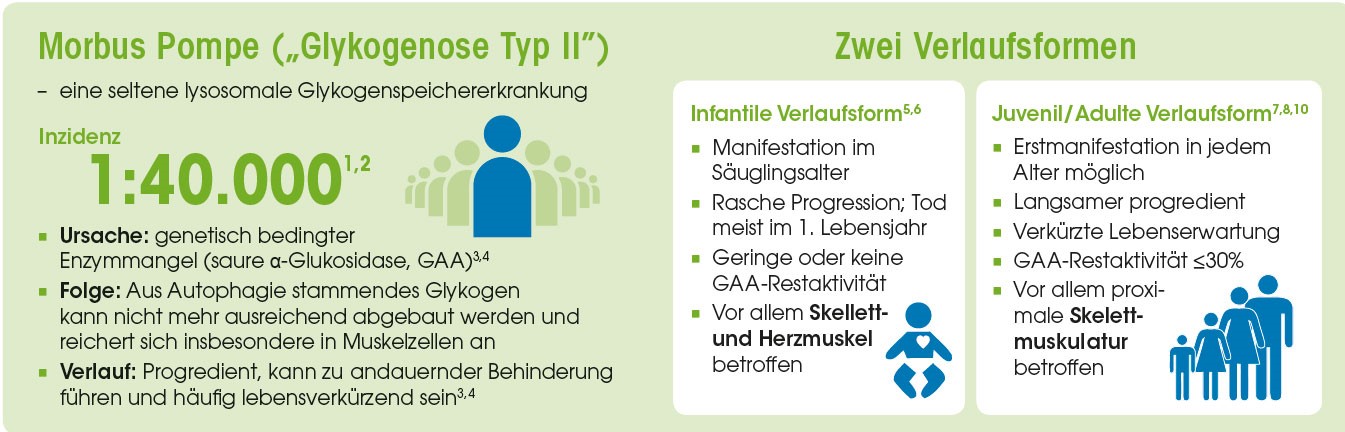
Pathophysiologisch kommt es beim M. Pompe aufgrund des Mangels des lysosomalen Enzyms saure alpha-Glukosidase zu einer zunehmenden Speicherung von Glykogen in den Lysosomen v. a. in den Muskelzellen und zu einer zunehmenden Schädigung des Muskelgewebes. Dabei kann neben der proximalen Skelettmuskulatur auch das Zwerchfell betroffen sein.
Häufige Schwierigkeit bei der Diagnostik insbesondere des Morbus Pompe: Aufgrund der Seltenheit und der unspezifischen Symptomatik kommen in der Differentialdiagnostik eine Reihe anderer neuromuskulärer Erkrankungen in Betracht. Dazu zählen unter anderem Gliedergürteldystrophien oder die Polymyositis.
Es empfiehlt sich, bei Hyper-CK-ämie und Atemschwäche auch an M. Pompe zu denken. Die "Kraftmessung des Zwerchfells" ist ein wichtiger und aussagekräftiger Parameter bei Pompe, u. a. messbar durch die FVC (forcierte Vitalkapazität) mit einem einfachen Lungenfunktionstest (Spirometer). Bei FVC < 80% des Sollwertes liegt ein Hinweis auf eine restriktive Ventilationsstörung vor, ein Abfall der FVC im Sitzen versus im Liegen um mehr als 20% deckt eine manifeste Zwerchfellschwäche auf.
Mehr über Morbus Pompe:
Die Genzyme GmbH, Deutschland, Teil der Sanofi Gruppe, unterstützt die Diagnostik-Initiative für lysosomale Speicherkrankheiten von Archimed Life Science GmbH. Daher kann Archimed Ärzten die Trockenblut-Testung kostenfrei anbieten. (Kontakthinweis: kostenfreie Service-Hotline zur Bestellung des Trockenblut-Tests 0800 /1115 200)
Wenn der Verdacht auf einen M. Pompe besteht, kann die Diagnose durch eine zuverlässige Aktivitätsmessung des Enzyms saure alpha-Glukosidase (GAA) per Trockenblut-Testung gestellt werden. Bei auffälligem Befund kann durch eine molekulargenetische Untersuchung aus der gleichen Trockenblutprobe die Diagnose gesichert werden. (mehr dazu HIER).
Für die kausale Therapie des Morbus Pompe steht seit 2006 Myozyme® als eine Enzymersatztherapie mit rekombinant hergestellter GAA zur Verfügung (mehr dazu HIER). Daneben ist es wichtig, symptomatische Therapiemöglichkeiten gemäß des individuellen Krankheitsbildes auszuschöpfen, dazu zählen zum Beispiel gezielte Physiotherapie und insbesondere auch eine frühzeitig abgestimmte Beatmungstherapie.
Nachfolgend stellte Prof. Cornelia Kornblum von der Klinik für Neurologie vom Universitätsklinikum Bonn unter dem Titel "Myositis und doch kein Effekt von Kortison?" den Fall einer vielgeplagten 62-jährigen Patientin vor.
Sie litt seit 3 Jahren unter Schmerzen im Schulter- und Beckengürtelbereich mit einer sich schleichend entwickelnden Muskelschwäche im Becken- und Oberschenkelbereich, was u.a. zu Problemen beim Treppensteigen führte. Dazu kamen verschiedene Gelenk- und gelegentlich auftretende Atembeschwerden nachts im Liegen, sodass sie mit hoch gelagertem Oberkörper schlief. Es wurde im Krankheitsverlauf eine Myositis (u. a. gestützt durch Muskelbiopsie-Befunde) diagnostiziert. Ein Muskel-MRT zeigte deutliche Auffälligkeiten (fettige Degeneration) v. a. der hinteren (dorsalen) Oberschenkelmuskeln beidseits. Bei weiterbestehender Diagnose Myositis verschlechterten sich die Beschwerden zunehmend trotz Therapie (u. a. mit Kortikosteroiden und Methotrexat) soweit, dass die Patientin nachts aufgrund der Atemstörungen Panikzustände hatte und eine belastungsabhängige Dyspnoe auch tagsüber auftrat. Mittlerweile schlief die Patientin nur noch im Sitzen und es stellte sich die Frage nach einer Angststörung. Neben der Belastungsintoleranz zeigte die Patientin eine Rumpfinstabilität und Probleme, aufrecht zu laufen. Weitere Untersuchungsergebnisse zeigten leicht erhöhte CK-Werte (350 -450 U/ml). Erst nach erneuter interdisziplinärer Begutachtung des Falls und erneuter Muskelbiopsie mit hinweisenden Befunden wurde der Verdacht auf einen Morbus Pompe geäußert. Biochemische Analysen aus dem Biopsiematerial zeigten eine verminderte Enzymaktivität der sauren alpha-Glukosidase und die genetische Untersuchung bestätigte die Diagnose schließlich. Die Enzymersatztherapie mit Myozyme® wurde eingeleitet.
Der Patientin geht es erfreulicherweise inzwischen wieder gut.
Frau Professor Kornblum betont, dass die Diagnose sicherlich schon früher hätte gestellt werden können und resümiert:
- Die Muskelbiopsie beim LOPD (Late On-set Pompe Disease) kann unauffällig oder nur unspezifisch verändert sein, insbesondere wenn sie nicht an der richtigen Stelle der betroffenen Muskulatur erfolgt!
- M. Pompe sollte früh differentialdiagnostisch bei Myalgien und Schwäche der Rumpf- und Gliedergürtelmuskulatur sowie Dyspnoe in Erwägung gezogen werden
- Eine milde bis moderate Hyper-CK-ämie sollte eine "red flag" sein, um die biochemische Analyse der GAA-Enzymaktivität einzuleiten: Ein Trockenblut-Test ist minimal invasiv, kostensparend und kann weitere invasive Diagnostik erübrigen.
Im Anschluss an die Fallvorstellungen fasste Herr Professor Benedikt Schoser, Leiter des neuromusklulären Zentrums am Friedrich Bauer-Institut in München, zusammen, wie wichtig eine frühe Diagnose des M. Pompe ist, denn v. a. die Probleme mit der Atmung führen zu frühzeitigem Versterben der unbehandelten Pompe-Patienten. Mit der Enzymersatztherapie steht eine wirksame, kausale Behandlungsmöglichkeit zur Verfügung. Eine aktuelle Metaanalyse1 zeigt für behandelte M. Pompe-Patienten einen deutlichen Überlebensvorteil und eine langfristige Stabilisierung und Verzögerung der Krankheitsprogression. Nach bereits zehn Jahren guter Erfahrung mit der Enzymersatztherapie (EET) hat das Europäische Pompe Consortium (EPOC) die Therapieerfolge bestätigt und Empfehlungen zur Behandlung von Pompe-Patienten für ganz Europa formuliert. Nach EPOC-Empfehlung sollte eine EET bei jedem symptomatischen Patienten mit bestätigter Diagnose begonnen werden.2
1Schoser B et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis., J Neurol 2017;264(4):621–630
2Van der Ploeg A et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience., Eur J Neurol 2017; 24(6):786-e31