- Arbeitsgemeinschaft pro Biosimilars. Handbuch Biosimilars. 2023. Link: https://probiosimilars.de/app/uploads/2022/12/ProBio-Handbuch-Biosimilars-2022-RZ-Web-DS.pdf. Letzter Zugriff: 28.09.2023
- Europäische Arzneimittel-Agentur (EMA) und Europäische Kommission. Biosimilars in der EU – Leitfaden für medizinische Fachkräfte. 2019. Link: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_de.pdf. Letzter Zugriff: 28.09.2023
- Bauer C et al. Bedarfsgerechte Versorgung mit modernen Biopharmazeutika nach 2022. 2021; Link: https://archiv.monitor-versorgungsforschung.de/efirst/Versorgung_Bios_2022/view.html. Letzter Zugriff: 09.10.2023
- Europäische Arzneimittel-Agentur (EMA). Ausschuss für Humanarzneimittel (CHMP). Guideline on similar biological medicinal products. 2014. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf. Letzter Zugriff: 28.09.2023
- Weise M et al. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191-3196.
Biosimilars: Regulierung, Zulassung, Besonderheiten
Die zunehmende Bedeutung biologischer Arzneimittel in der modernen Medizin hat zu einer wachsenden Nachfrage nach kostengünstigeren Alternativen geführt. In diesem Kontext haben Biosimilars in den vergangenen Jahren stark an Bedeutung gewonnen.<sup>1</sup>
Die zunehmende Bedeutung biologischer Arzneimittel in der modernen Medizin hat zu einer wachsenden Nachfrage nach kostengünstigeren Alternativen geführt. In diesem Kontext haben Biosimilars als vielversprechende Option in den vergangenen Jahren stark an Bedeutung gewonnen.1 Grund genug also, sich mit diesem Thema auseinanderzusetzen. Eine neue eCME ermöglicht Ihnen jetzt einen umfassenden Überblick zu allen Fragen rund um Biosimilars: Was ist der Unterschied zwischen Biosimilars und Generika? Was sind Besonderheiten bei Entwicklung und Zulassung von Biosimilars? Oder: Was versteht man unter Mikroheterogenität oder Extrapolation?
Biosimilars: Ähnlich aber nicht identisch
Biosimilars sind ähnliche, aber nicht identische Folgeprodukte biologischer Arzneimittel (Biologika), die aber genauso verträglich und wirksam sind wie das Referenzarzneimittel.2 Im Gegensatz dazu sind Generika eine chemisch identische Kopie des Originalarzneimittels. Anders als bei Generika, reichen für die Zulassung von Biosimilars deshalb einfache Bioäquivalenz-Studien nicht aus.2 Es bedarf umfangreicher präklinischer und klinischer Studien, um die Vergleichbarkeit eines Biosimilars nachzuweisen.2 Dadurch wird sichergestellt, dass sich ein Biosimilar von seinem Referenzarzneimittel nicht stärker unterscheidet als einzelne Chargen des Referenzarzneimittels untereinander.1,2 Die Entwicklung und Zulassung eines Biosimilars ist dadurch deutlich aufwendiger als die eines Generikums: Es werden etwa acht Jahre und bis zu 200 Millionen USD benötigt, bis ein Biosimilar die Marktreife erreicht – bei einem Generikum sind es im Schnitt nur 5 Millionen USD.3
Entwicklung und Zulassung eines Biosimilars
Grund für die im Vergleich zu Generika um ein Vielfaches höheren Entwicklungskosten sind strenge wissenschaftliche und regulatorische Anforderungen, die die EMA vor eine Zulassung durch die Europäische Kommission gestellt hat, um die Vergleichbarkeit, Qualität, Wirksamkeit und Verträglichkeit von Biosimilars sicherzustellen (siehe Abbildung).2 Im Gegensatz zu Referenzarzneimitteln liegt der Schwerpunkt bei der Entwicklung von Biosimilars auf Studien zur pharmazeutischen Qualität und nicht-klinischen Studien.2 Während sich die Anforderungen an die Prüfung der pharmazeutischen Qualität nicht von denen der Erstanbieter-Präparate unterscheiden, können klinische Untersuchungsprogramme verkürzt werden. So können beispielsweise Phase-II-Studien zur Dosisfindung oft entfallen.2
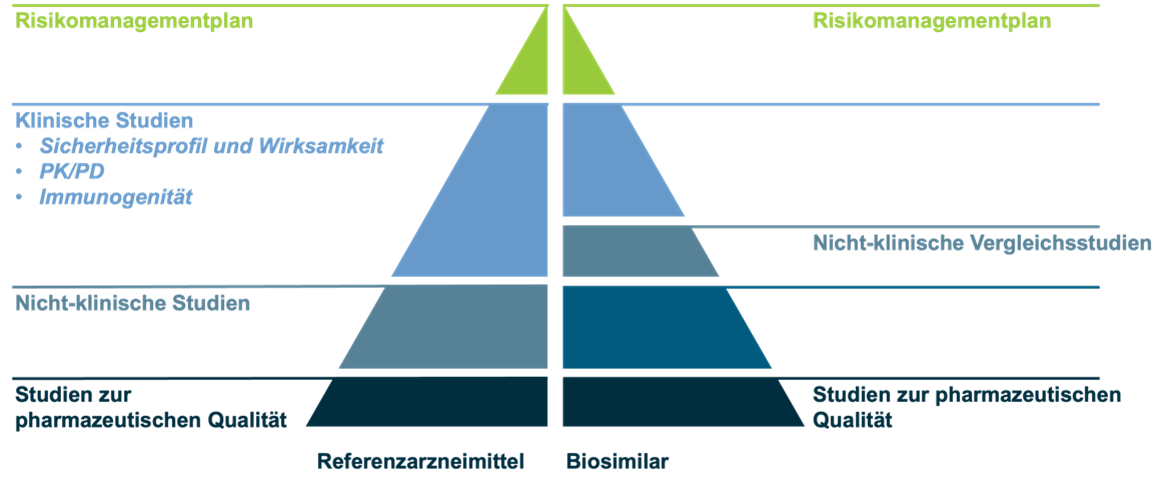
Anforderung an eine Zulassung eines Biosimilars bzw. eines Referenzarzneimittels in der EU (Abbildung modifiziert nach Europäische Arzneimittel-Agentur (EMA) und Europäische Kommission. Biosimilars in der EU – Leitfaden für medizinische Fachkräfte. 2019.2)
Eine weitere Besonderheit bei der Zulassung von Biosimilars ist, dass die Vergleichbarkeit lediglich in einem der Hauptanwendungsgebiete des Referenz-Biologikums nachgewiesen werden muss.2,4 Die Zulassung erfolgt dann für alle Anwendungsgebiete des Referenzarzneimittels. Diese sogenannte Extrapolation der Anwendungsgebiete durch die EMA ist möglich, wenn die Gesamtheit der Evidenz dies auch unterstützt, das Biosimilar also in allen klinischen und nicht-klinischen Studien seine Vergleichbarkeit demonstrieren konnte.2,5 Dies verringert nicht nur die Zahl notwendiger klinischer Studien, sondern ermöglicht es, das Biosimilar rasch bei einem breiten Patientenspektrum einsetzen zu können.
Online-Fortbildung zum Thema Biosimilars
Sie wollen tiefer in die Thematik eintauchen? Mehr über Biosimilars, Mikroheterogenität bei biologischen Arzneimitteln oder die Extrapolation von klinischen Daten erfahren Sie jetzt auch in der eCME "Biosimilars: Ein etabliertes Konzept und praktische Erfahrungen aus der Anwendung". In dieser Fortbildung geht es außerdem um klinische Erfahrungen mit Biosimilars und die Sicherheitseinstufung von Biosimilars der EMA. Zudem erhalten Sie praktische Tipps zur Kommunikation mit Patienten und zur Minimierung von Nocebo-Effekten bei der Umstellung auf Biosimilars. Nutzen Sie diese Gelegenheit, um Ihr Wissen über Biosimilars zu erweitern.
Hier finden Sie weitere Informationen zur eCME "Biosimilars: Ein etabliertes Konzept und praktische Erfahrungen aus der Anwendung" und haben die Möglichkeit, sich direkt für diese Fortbildung anzumelden.
- Weitere Hintergrundinformationen zum Thema Biosimilars finden Sie in diesem Beitrag.
- Einen Videobeitrag zum Thema finden Sie hier.
Referenz
Biogen-221349 v1.0 09/2023